近日,我校化学系龚隽一副教授与香港中文大学(深圳)唐本忠院士团队合作,在材料计算与人工智能交叉领域取得重要进展。研究团队开发出一套基于立方采样的电子密度生成卷积残差网络(GED-CRN)的模型,仅使用19个训练分子即实现了接近MP2理论级别的高精度电子密度预测,为数据稀缺体系(如聚集诱导发光材料)的性质预测提供了新思路。该研究成果发表于国际知名期刊《Aggregate》(Aggregate 2025, e70119)。
电子密度分布是理解分子光电性质与反应机制的核心物理量,传统量子化学计算方法虽精度高但耗时巨大,尤其在处理聚集态体系时往往“算不动”。机器学习方法虽可加速计算,却严重依赖大量高质量数据,而功能材料研究中往往面临“数据零散、样本稀缺”的现实困境。
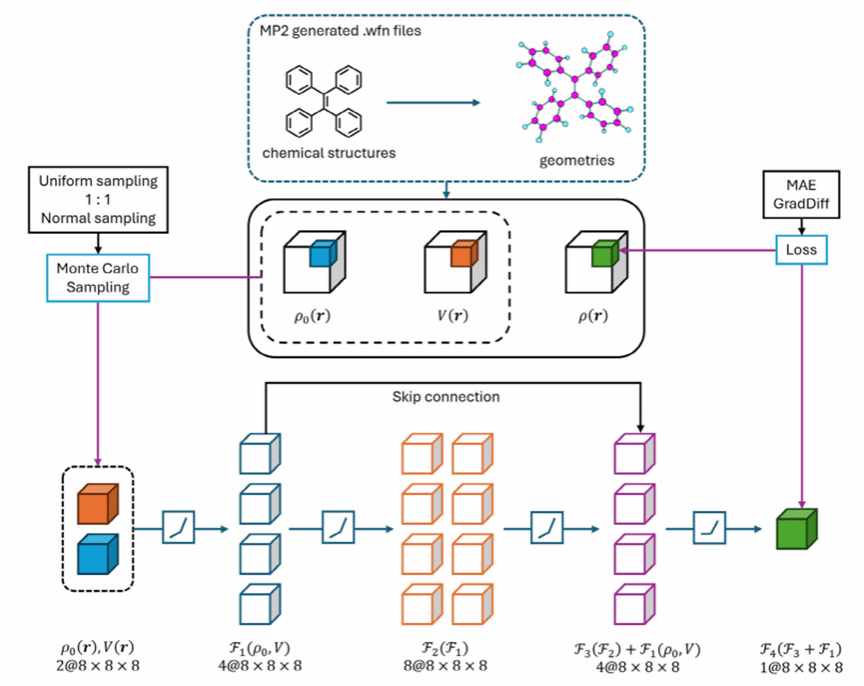
针对这一难题,研究团队提出了一种融合物理先验与深度学习的小样本学习框架。通过空间立方采样策略,将每个分子转换为数千个有效训练样本,极大提升了数据利用效率。模型采用双通道输入机制,结合电子密度与核电势场信息,引入3D卷积残差网络与梯度差异损失函数(GradDiff),实现了电子密度及其梯度分布的高保真预测。
结果表明,GED-CRN在QM9通用分子库和ASBase发光材料数据库测试中表现优异,整体平均绝对误差低于3.0×10-4 bohr-3,对聚集诱导发光(AIE)体系误差更降低50%。单分子电子密度预测仅需秒级完成,较传统方法提速三个数量级,显著提升了发光材料的虚拟筛选效率。
该研究不仅为AIE机制研究与材料设计提供了新工具,也展示了“少样本学习”在计算化学中的应用潜力。团队已公开全部模型代码,并正推进激发态版本的开发,推动量子机器学习在材料科学中的深度应用。
龚隽一博士现任我校化学系预聘副教授,长期从事发光材料的理论模拟与智能设计研究,在Adv. Sci.、Adv. Funct. Mater.、Chem. Sci.等期刊发表论文近50篇,H-index达29。